ساختار میتوکندری در انسان و نقش آن در ایجاد بیماری های مختلف میتوکندری
8,900 تومانشناسه فایل: 4780
مقدمه
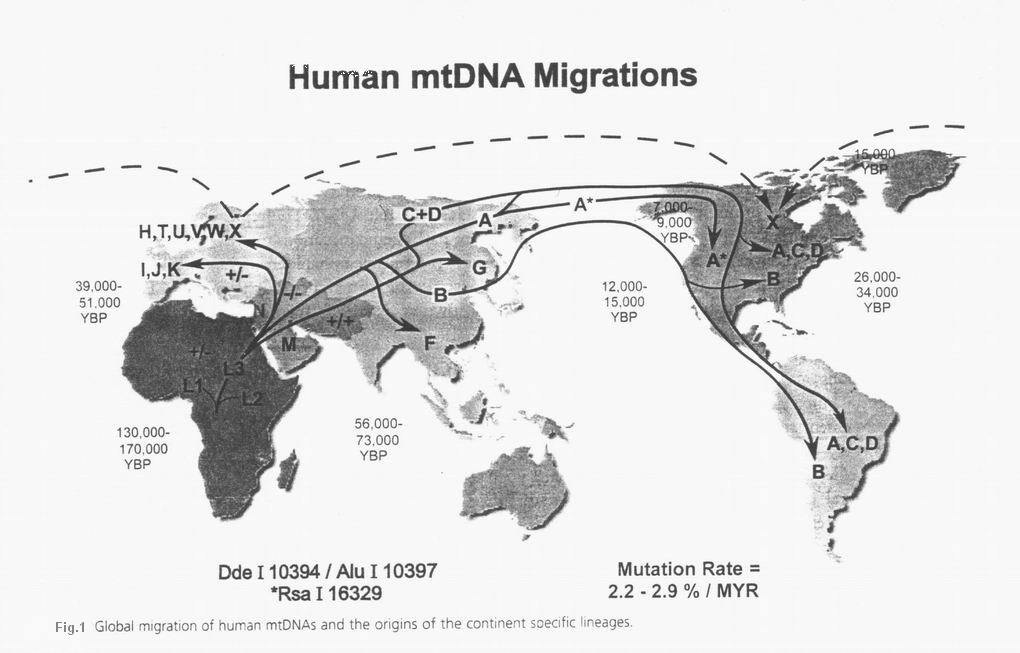
اولین گزارشات در ارتباط با ساختارهای درون سلولی شبه میتوکندری به 150 سال پیش برمیگردد. واژه میتوکندری که از دو کلمه یونانی mitos بمعنی نخ یا رشته و chondros به معنی گرانول منشا گرفته است؛ برای اولین بار صد سال پیش مورد استفاده قرار گرفت. عملکرد اصلی این ارگانل کروی یا میلهای شکل که صدها عدد از آن در یک سلول وجود دارد، فسفریلاسیون اکسیداتیو است؛ به عبارت دیگر اکسیداسیون سوبستراها به Co2 و آب و فراهم کردن ترکیب پر انرژی ATP برای سلولها؛ و به همین دلیل است که میتوکندری را نیروگاه یا موتورخانه سلول نیز مینامند. بیماریهای دژنراتیو بسیار زیادی تا به امروز با نارساییها و اختلالات میتوکندری مرتبط شدهاند. این بیماریها میتوانند در اثر موتاسیون در DNA میتوکندری و یا DNA هسته ایجاد شوند. اولین بیماریهای میتوکندریایی که در سطح مولکولی درک شدند؛ در یک بیمار CPEO (فلج مزمن پیشرونده عضلات چشمی خارجی) و KSS (سندرمkearns-sayre) گزارش شدند. در همان زمان wallace موتاسیونی نقطهای را در ژن ND6 گزارش کرد که با LHON (نوروپاتی چشمی ارثی لبر) مرتبط است. در سال 1990، دوموتاسیون جدید، یکی در ژن لایزیل- tRNA در سندرم MERRF و دیگری در ژن لوسیل – tRNA در سندرم MELAS گزارش شدند. طیف فتوتیپی بیماریهای میتوکندریایی از میوپاتیهای نادر تا بیماریهای متعدد را شامل میشود. برخی موتاسیونهای mtDNA، علائم و نشانههای منحصر و ویژهای دارند؛ مثل جهشهای اشتباهی که موجب نوروپاتی چشمی ارثی لبر میشوند در حالیکه بقیه تظاهرات مولتی سیستم متنوعی را شامل میشوند مثل جهشهای حذفی که موجب CPEO میشوند. بیماریهای میتوکندریایی بواسطه وراثت مادری، وراثت منرلی و نیز نوترکیبیهای دوتایی نو، قادر به انتقال میباشند. این پیچیدگی ژنتیکی از این حقیقت ناشی میشود که میتوکندری از حدود 1000 ژن که در بین ژنوم میتوکندری و هسته پخش شدهاند، تشکیل شده است. علاوه بر این بیماریهای میتوکندریایی غالباً شروع تاخیری و یک دوره پیش رونده دارند که احتمالاً از تجمع جهشهای سوماتیک mtDNA در بافتهای post-mitotic حاصل شدهاند. این موتاسیونهای سوماتیک mtDNA همچنین در سرطان و پیری نیز نقش دارند. اگرچه بیماریهای میتوکندریایی هر ارگانی را ممکن است درگیر کنند اما این بیماریها غالباً CNS، عضلات اسکلتی، قلب، کلیه و سیستمهای اندوکرین را تحت تاثیر قرار میدهند. علت این پیچیدگیهای فتوتیپی، نقش مهم میتوکندری در انواع پروسههای سلولی شامل تولید انرژی سلولی بوسیله فسفریلاسیون اکسیداتیو، تولید گونههای سمی فعال اکسیژن (ROS) به عنوان یک محصول جانبی در فسفریلاسیون اکسیداتیو و تنظیم شروع آپوپتوزاز طریق فعال شدن نفوذپذیری پورهای انتقالی میتوکندری (mtPTP) است.
کلیات
ساختار میتوکندری
میتوکندری واجد یک غشای بیرونی و یک غشای داخلی است که دو فضای داخلی را ایجاد میکنند: ماتریس داخلی و فضای بین دو غشا که بسیار باریک است. غشای داخلی چینخورده و تعداد زیادی کریستا ایجاد میکند که کل سطح آنرا به مقدار زیادی افزایش میدهد. سطح وسیع غشای داخلی، آنزیمهای دستگاه مولد انرژی میتوکندریایی (زنجیره تنفسی) را در خود جای داده است. ماتریکس میتوکندری واجد نسخههای یکسان متعددی از ژنوم میتوکندری، ریبوزومهای ویژه میتوکندری (میتو ریبوزوم)، tRNAها و آنزیمهای متنوعی است که برای بیان ژنهای میتوکندری مورد نیازند. (20)
ژنوم میتوکندری انسان
حضور DNA در میتوکندری در سال 1963 و با استفاده از میکروسکوپ الکترونی مشخص شده است. DNA میتوکندریایی انسان یک مولکول مدور بسته دو رشتهای با 16569 جفت نوکلئوتید است. دو رشته mtDNA که به رشتههای H (سنگین) و L (سبک) معروفند، یک عدم تقارن غیر معمول در ترکیب بازی هایشان دارند. زنجیره H غنی از پورین است در حالیکه زنجیره L غنی از پیریمیدین میباشد. سبک و سنگین به تحرک متفاوت رشتهها در گرادیانهای سزیم کلرید قلیایی اطلاق میشود. mtDNA انسان یکی از متراکمترین و فشردهترین بخشهای اطلاعات ژنتیکی است. در mtDNA، اینترون وجود ندارد و حتی بعضی از ژنهای آن همپوشانی دارند. DNA میتوکندریایی انسان واجد ژن هایی برای سیزده پروتئین (که همگی زیر واحدهای کمپلکسهای آنزیمی زنجیره تنفسی هستند)، 22 tRNA و دو rRNA است.
نوترکیبی mtDNA
از سالها قبل شواهدی مبنی بر اینکه مخلوط شدن mtDNA در داخل سلولهای سوماتیک ممکن است نوترکیبی ایجاد کند، وجود داشته است. هر چند تعداد دفعات نوترکیبی در سلولهای کشت شده پستانداران کم است، اما نوترکیبیهای درون ملکولی ممکن است مکرراً رخ دهند . اگر یک mtDNA واجد حذف به سلول زایای موش ماده وارد شود، منجر به ایجاد استرینی از موش میشود که در آن فرزندان واجد mtDNAهای نرمال و واجد حذف هستند. مولکول های دوپلیکیت نیز افزایش مییابند که به نظر میرسد از ترکیب مولکول های نرمال و واجد حذف بوجود آمده باشند. با این حال هیچ مشاهدهای مبنی بر وجود نوترکیبی در بین ردههای مختلف mtDNA انسانی وجود ندارد. بنابراین احتمالاً در بین ردههای mtDNA انسانی، نوترکیبی رخ نمیدهد.
نتیجه گیری
آنچه تاکنون مورد بررسی قرار گرفته و روشن شده است، حکایت از آن دارد که میتوکندری واجد نقشی اساسی در بیماریهای دژنراتیو، سرطان و پیری است. این اثرات مختلف از طریق حالت رداکس سلولی که توسط میتوکندری و از طریق اکسیداسیون و احیای NADH,H+ و NAD+ حفظ می شود؛ با همدیگر مرتبط هستند.
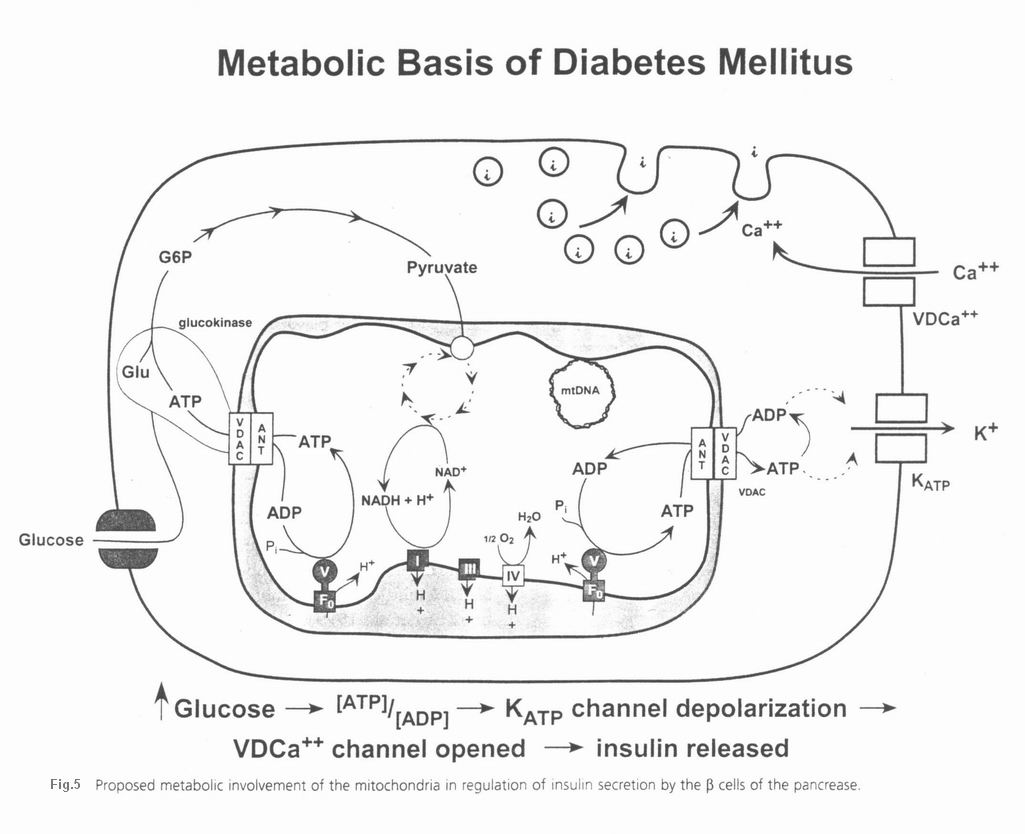
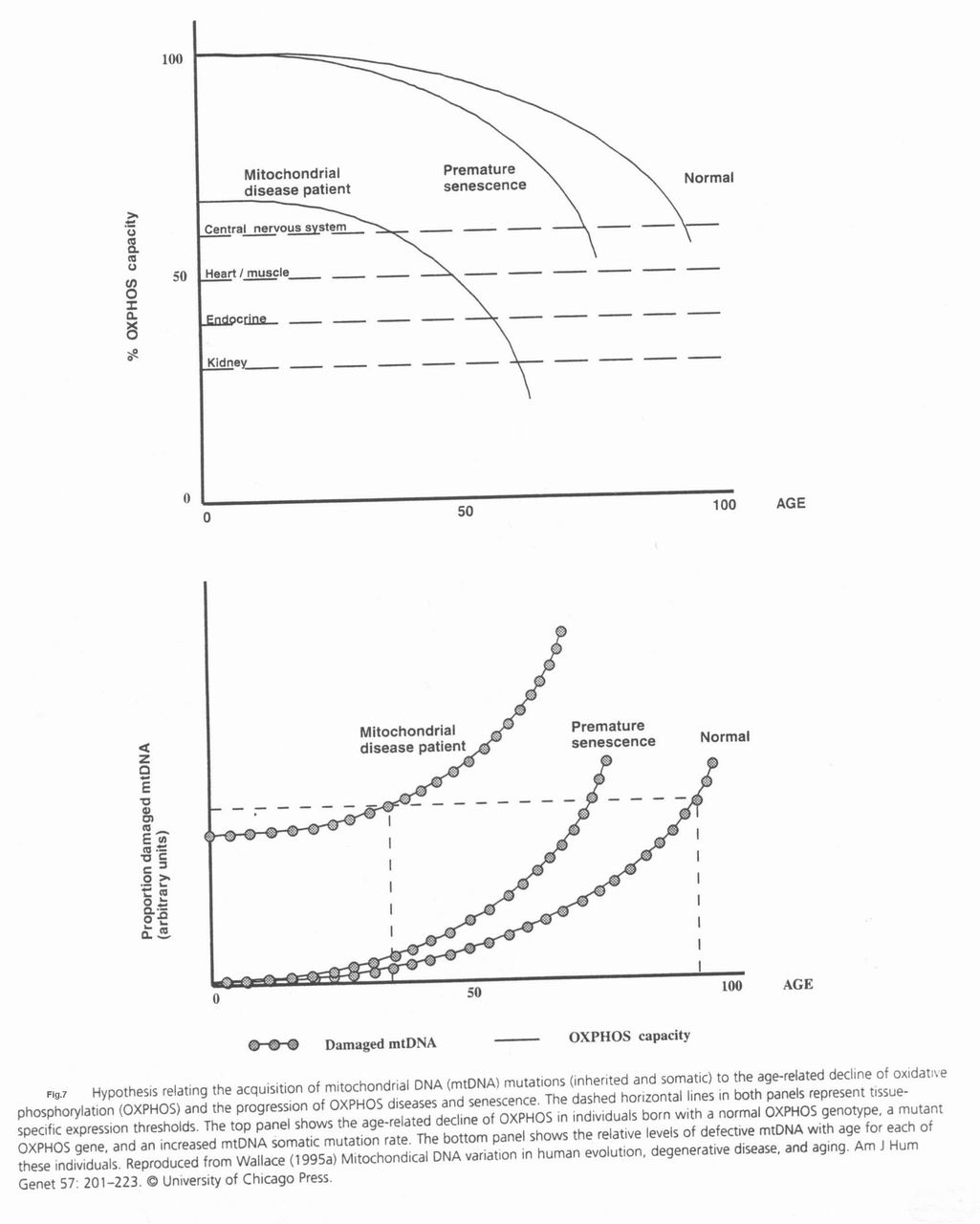
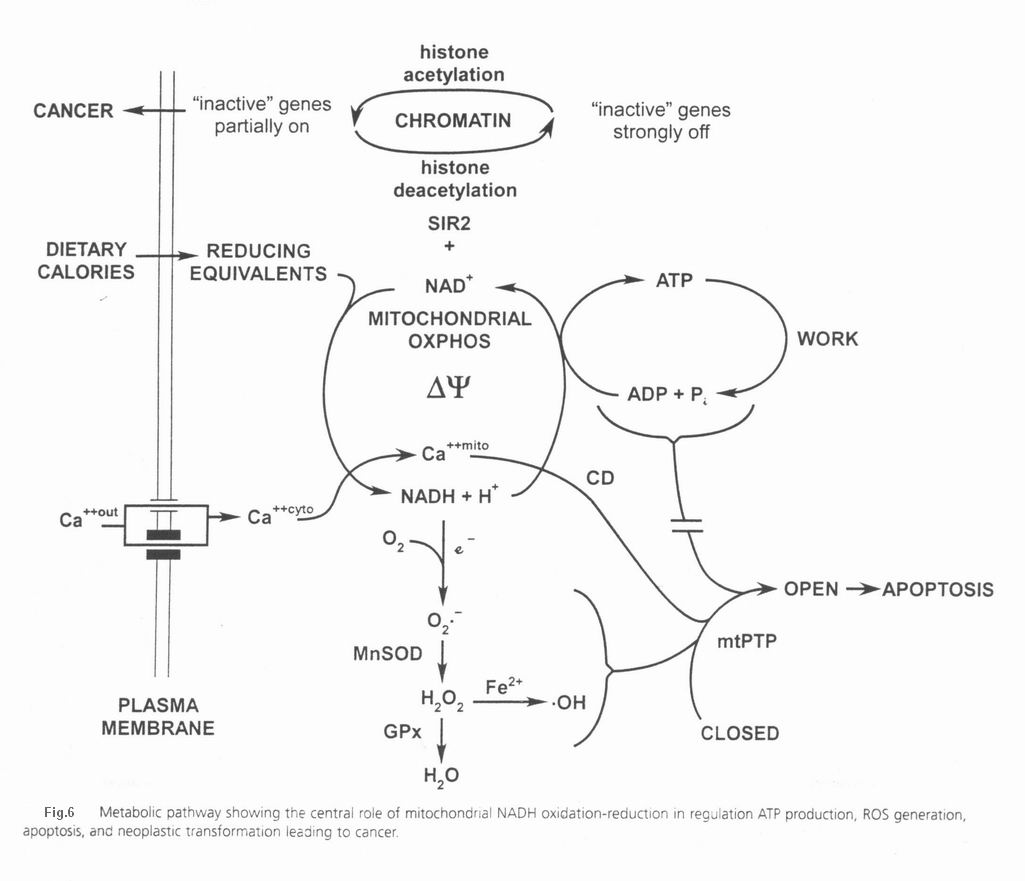
انرژی غذایی وارد میتوکندری شده و اکی والان های احیا را جهت احیای NAD+ به NADH,H+ تامین می کند. سپس NADH,H+ مجدداً توسط میتوکندری اکسید شده و ایجاد می کند. حاصله صرف سنتز ATP از ADP و Pi شده و یا صرف جذب کاتیون هایی مثل می شود. موقعی که فعالیت سلول بالاست، ATP به طور فعال هیدرولیز شده و موجب افزایش ADP سلول می شود که توسط ANT به داخل ماتریکس منتقل می شود. ADP افزایش یافته در ماتریکس، مجدداً و به خرج فسفریله می شود که این عمل با اکسیداسیون NADH,H+ به NAD+ توسط زنجیره انتقال الکترون همراه است . هنگامی که انرژی غذایی از میزان لازم جهت کار سلول بیشتر می شود، تمامی ADP به ATP فسفریله می شوند، هایپر پلاریزه می شود و NAD+ تدریجاً به NADH,H+ احیا می شود. زیاد شدن اکیولانهای احیای NADH ، موجب احیای ETC می شود. که این امر انتقال الکترون به و ایجاد را تحریک میکند. آنیون سوپراکساید میتوکندریایی با غشاها، پروتئین ها و DNA میتوکندریایی واکنش داده و موجب آسیب آنها می شود. همچنین توسط MnSOD میتوکندریایی به تبدیل می شود. به هسته نشت پیدا کرده و سبب القای موتاسیون در nDNA شده و پروتئین PARP را جهت شروع تخریب NAD+ فعال می کند. علاوه بر این تجمع موتاسیونهای سوماتیک mtDNA نیز زنجیره انتقال الکترون را مهار کرده و تولید ROS را تحریک می کند. همچنین آسیب غشای پلاسمایی یا تحریک گیرندههای NMDA نورونها توسط گلوتامات، کلسیم سیتوزولی را افزایش داده که متعاقباً افزایش غلظت آن در میتوکندری را به دنبال دارد. کلسیم اضافی به سیکلوفیلین D متصل شده؛ استرس اکسیداتیو را افزایش داده و را کاهش می دهد که همگی به mtPTP اثر کرده و در نهایت موجب نفوذپذیری و از دست رفتن سلول در نتیجه آپوپتوز می شوند. این از دست رفتن سلولی موجب نقص و از بین رفتن بافت و ارگان و نارسایی سیستمیک می شود. کاهش NAD+ سلول چه از طریق احیای آن به NADH,H+ و چه تخریب توسط PARP، موجب مهار پروتئین خاموش کننده کروماتین هسته ای SIR2 می شود. Nuclear chromatin silencing protein با استفاده از NAD+ بعنوان سوبسترا، گروههای استیل هیستونها را شکسته و بنابراین ژنهای «off» را غیر فعال نگه می دارد. ژنهای off ژنهایی هستند که رونویسی آنها قدغن بوده و در حالت عادی خاموش و غیر فعالند مثل انکوژن ها. در غیاب SIR2 فعال ، هیستون های نوکلئوتومی به میزان زیادی استیله شده و موجب رونویسی غیر مجاز ژن هایی می شود که در حالت طبیعی نباید رونویسی شوند( ژنهای off)؛ فرایندی که ویژگی خاص بافت های پیر است. این فرایند نه تنها پروتئین های ساختاری را فعال می کند بلکه قادر است پروتو انکوژن های غیرفعال را نیز فعال نماید. این فعال شدن رونویسی ، تغییر پروتوانکوژنها در اثر به تدریج احتمال ایجاد سرطان را افزایش می دهد. اکنون این مدل توضیح می دهد که چرا محدودیت های کالریک (تغذیه ای) نه تنها طول عمر (Longevity) را افزایش می دهد بلکه ریسک سرطان را نیز کاهش می دهد. با کاهش دریافت کالری و تعادل اکیولانهای احیا با هیدرولیز ATP وابسته به کار و فعالیت، NAD+ به فرم اکسید باقی خواهد ماند. به عبارت دیگر هر چه فعالیت بیشتر باشد، هیدرولیز ATP بیشتر بوده و بنابراین تبدیل ADP به ATP نیز افزایش می یابد که این مهم مستلزم تبدیل اکیولانهای احیا به اکسید در ETC است. این عمل ایجاد الکترونهای مازاد و تولید ROS ناشی از آن را برطرف کرده و بنابراین استرس اکسیداتیو، ایجاد جهش در mtDNA، از دست رفتن سلول توسط آپوپتوز و آسیب DNA هسته ای و فعال شدن آن توسط را به حداقل می رساند. همچنین حفظ ذخیره NAD+ ،SIR2 را کاملاً فعال نگه داشته و بنابراین فعال شدن انکوژنها را ساپرس می کند. بنابراین بیماری میتوکندریایی و نقش میتوکندری در سرطان و پیری را می توان در نتیجه اثر متقابل دو فاکتور ژنتیکی میتوکندریایی در نظر گرفت: 1- به ارث رسیدن موتاسیونهای مضر mtDNA یا nDNA در یک ژن میتوکندری 2- تجمع وابسته به سن موتاسیونهای سوماتیک mtDNA که موجب نقص میتوکندری ، افزایش تولید ROS و آپوپتوز می شوند. هر شخص با آرایشی از الل های mtDNA و nDNA که ظرفیت بیوانرژتیک آغازین او را تعیین می کند، متولد می شود. اگر فردی یک ژنوتیپ انرژتیک به ارث ببرد او ظرفیت انرژی آغازین بالایی را دارا خواهد بود که از حداقل آستانه انرژتیک مورد نیاز بافتهایش ، بیشتر است. چنانچه فردی یک موتاسیون مضر به ارث ببرد، ظرفیت انرژتیک آغازین او پایین و تولید ROS بالا خواهد بود. همچنانکه سن فرد افزایش می یابد، موتاسیون های سوماتیک mtDNA در سلولهای post-mitotic تجمع یافته و تولید انرژی بافتی او کمتر و میزان تولید ROS بیشتر می شود. در نهایت اینکه نقایص میتوکندریایی ارثی و سوماتیک در مجموع ظرفیت انرژتیک بافتی را از آستانه بیوانرژتیک پایین تر آورده و موجب آپوپتوز و نارسایی ارگانی و نیز با فعال کردن پروتوانکوژنهای nDNA موجب سرطان می شوند.
فهرست مطالب و منابع
- مقدمه
- ساختار میتوکندری
- ژنوم میتوکندری انسان
- میتو کندریها نیمه خود مختار هستند
- میتو کندریها وراثت مادری دارند
- هتروپلاسمی و تفکیک رپلیکاتیو
- نوترکیبی mtDNA
- کامل شدن mtDNA
- میزان بالای موتاسیون در mtDNA
- تنوع پلی مورفیک mtDNA در جمعیتهای انسانی
- ژنتیک میتوکندری (همانندسازی، رونویسی و ترجمه mtDNA)
- فرایندهای میتوکندریایی
- میتوکندری و پاسخ به استرس
- بیان آستانهای
- بیماریهای میتوکندریایی ناشی از جهشهای سیستمیک
- LHON (نوروپاتی چشمی ارثی لبر)
- مکانیسمهای پاتو فیزیولوژیکی احتمالی LHON
- LHON، مولتیپل اسکلروزیس و دیستونی
- بیماری پارکینسون (PD) و بیماری هانتینگتون (HD)
- ژنتیک کروموزومی بیماری پارکینسون
- جهشهای mtDNA در PD
- اختلالات میتوکندریایی در PD
- نارساییهای میتوکندریایی در بیماری هانتینگتون
- رتینیت پیگمنتوزا (RP) و سندرم لی (LS)
- موتاسیونهای mtDNA در RP و سندرم لی
- میوپاتی و انسفالومیوپاتیهای میتوکندریایی
- ضعف عضلانی پیشرونده و مرتبط پا موتاسیونهای سیتوکروم mtDNA b
- انسفالومیوپاتیهای ناشی از جهشهای ژن COX mtDNA
- میوپاتیهای میتوکندریایی ناشی از موناسیونهای TRNA ژنوم میتوکندری
- کاردیو میوپاتیهایپرتروفیک و میوپاتی ناشی از جهشهای mtDNA
- انسفالومیوپاتیهای ناشی از جهشهای mtDNA
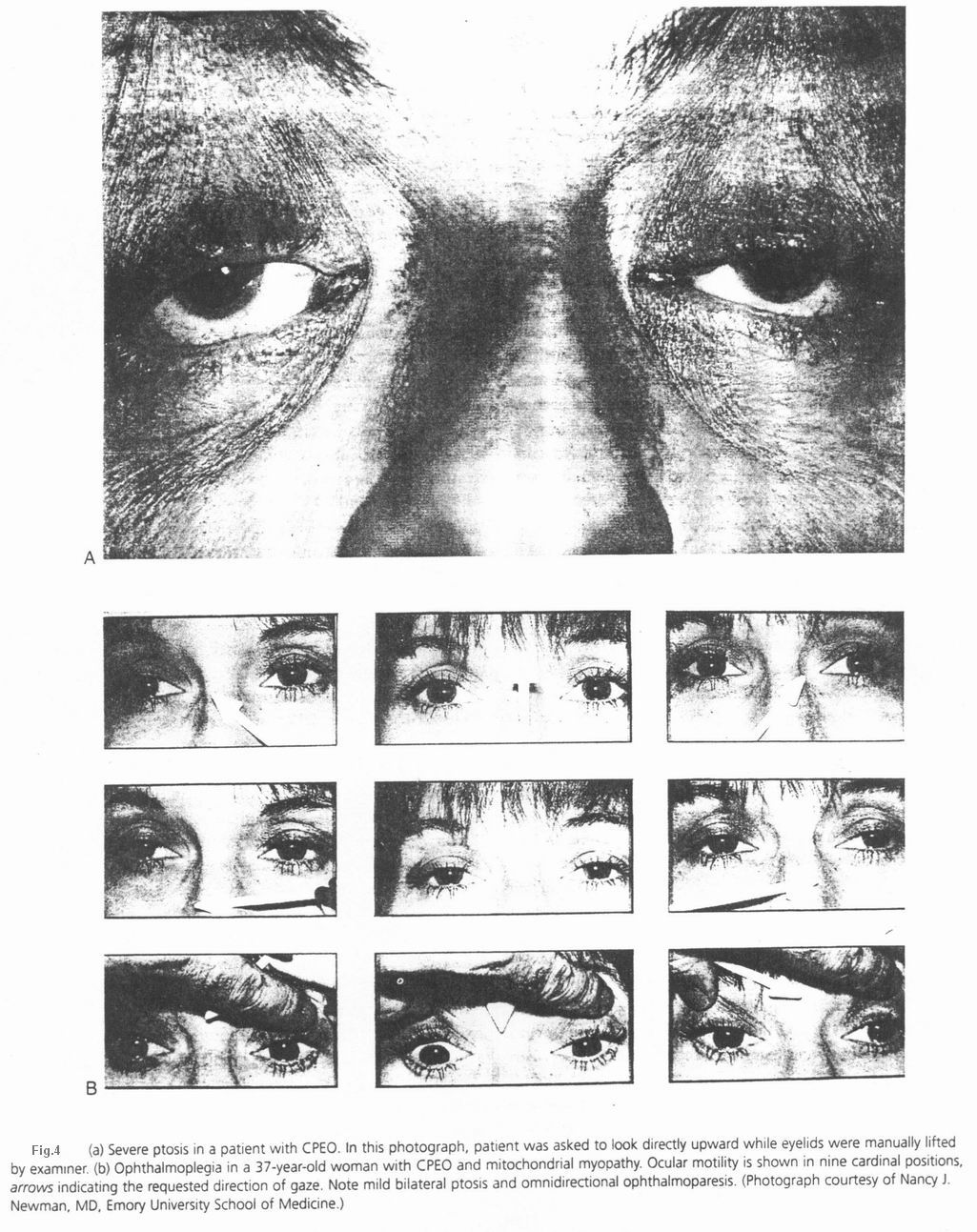
- افتالموپلژیا، پتوزیس و میوپاتی میتوکندریایی
- افتالموپلژیای ناشی از جهشهای mtDNA
- CPEO و KSS مرتبط با موتاسیونهای نوآرایی mtDNA
- CPEO ناشی از موتاسیونهای تعویض باز mtDNA
- سندرم مغز استخوانی پانکراسی پیرسون
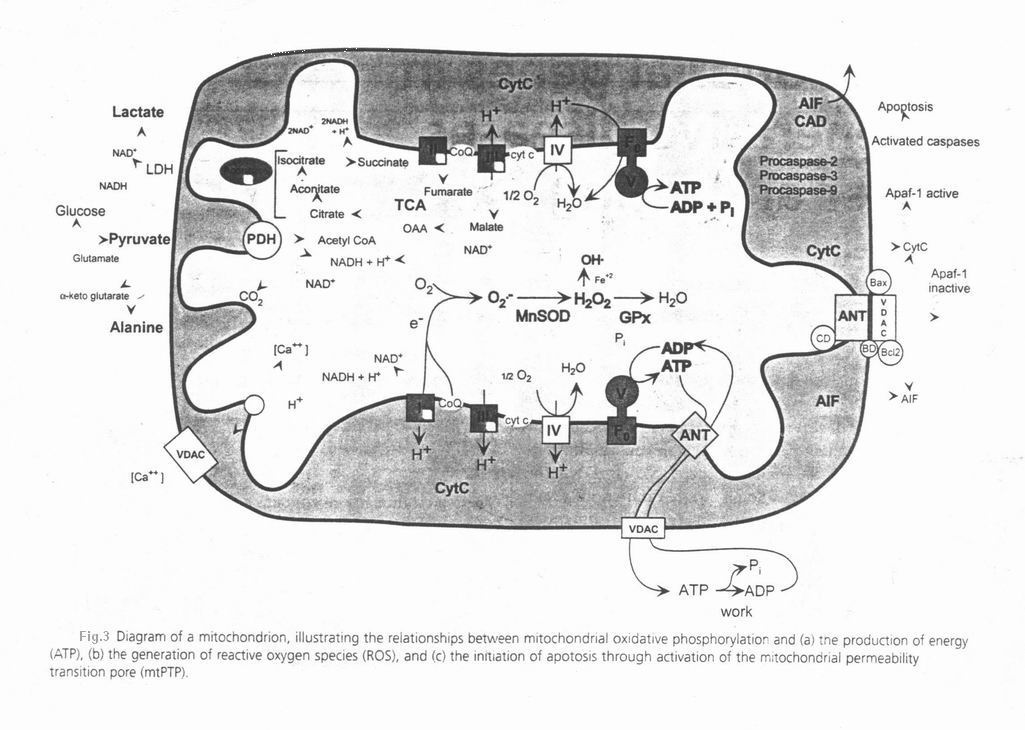
- دیابت ملیتوس
- تیپ II دیابت ملیتوس بوسیله نوآراییهای (حذفها و دوپلیکاسیون ها) mtDNA
- ایجاد میشود
- دیابت تیپ II ناشی از موتاسیونهای تعویض باز mtDNA
- میوپاتی و دیابت
- پاتوفیزیولوژی دیابت و کری
- کری به ارث رسیده از مادر و یا کری القا شده توسط آمینوگلیکوزید
- دمانس به عنوان یک بیماری میتوکندریایی
- بیولوژی و ژنتیک بیماری آلزایمر
- اختلالات میتوکندریایی در AD
- بیماری آلزایمر ناشی از جهشهای mtDNA
- دیس کندروپلازی متافیزیک یا هیپوپلازی مویی- غضروفی ناشی از جهشهای
- RNASE MRP
- بیماریهای مولتی فاکتوریال و mtDNA
- جهشهای سوماتیک mtDNA در بیماریهای دژنراتیو، سرطان و پیری
- تجمع جهشهای سوماتیک mtDNA مرتبط با سن
- آنمی سیدروبلاستیک ایدیوپاتیک
- بیماری ایسکمی قلبی و کاردیومیوپاتی اتساعی
- بیماریهای نورودژنراتیو؛ HD, PD و AD
- بیماری پارکینسون و بیماری هانتینگتون
- بیماری آلزایمر
- موتاسیونهای سوماتیک mtDNA در دیگر بیماریهای کمپلکس
- موتاسیونهای سوماتیک در سرطان
- نتیجهگیری
- منابع
منابع
- Allen J.C., Mikhailovsky I.E., Sukernik R.I., Wallace D.C.; The role of mtDNA background in disease expression: a new primary LHON mutation associated with western eurasian haplogroup J. Hum Genet; 2002; 110(2): 130-138.
- Alcolado J.C., thomas A.W.; Maternally inherited diabetes mellitus: the role of mitochondrial DNA defects. Diabet Med; 1995;12:102-108.
- Brown M.D., Voljavec A.S., latt M.T., Wallace D.C.; Leber’s hereditary optic neuropathy: A model for mitochondrial neurodegenerative disease. FASEB.J; 1992b; 6: 2791-2799.
- Bianca J.C., Rene F.M., Jeroen G., Marianne d.,Hubert J.;Mutation analysis of the entire mitochondrial genome using denaturing high performance liquid chromatography. Nucleic Acids Research; 2000; Vol. 28; No.20 : 89.
- Bruno C.,Martinuzzi A., Tang y.;A stop-codon mutation in the human mtDNA cytochrome C oxidase I gene disrupts the functional structure of complex IV. Am.J. Hum. Genet.; 1999; 65:611-620.
- Brown M.D., Wallace D.C.; Molecular basis of mtDNA disease. J.Bioenerg. Biomembr; 1994a; 26: 273-289.
- Brown M.D., Shoffner J.M., Kim Y.L.; Mitochondrial DNA sequence analysis of four Alzheimer’s and parkinson’s disease patients. Am.J. Hum. Genet; 1996; 61: 283-289.
- Bindoff L.A., Birch M.,parker W.D., Turnbull D.M.; Mitochondrial function in parkinson’s disease. Lancet; 1989;2:49.
- Brockington M.,Al Sanjari N.,Sweeney M.G.,Harding A.E.; kearns-sayre syndrome associated with mitochondrial DNA deletion or duplication:A molecular genetic and pathological study. J.Neurol. sci; 1995; 131: 78-87.
- Brennan W.A., Bird E.D., Aprile J.R.; Regional mitochondrial respiratory activity in Huntington’s disease brain. J.Neurochem.; 1985;44: 1948-1950.
- Bindoff L.R., Howell N.,Poulton J.; Abnormal RNA processing associated With a novel tRNA mutation in mitochondrial DNA:A potential disease mechanism. J.Biol. chem.; 1993;268 : 19559-564.
- Blin O.,Desnuelle C., Rascol O.; Mitochondrial respiratory failure in skeletal muscle from patients with parkinson’s disease and multiple system atrophy. J. Neurol. Sci.; 1994; 125:95-101.
- Bofol D., Scacco S.C., Solarino G., Papa s.; Decline with age of the respiratory chain activity in human skeletal muscle. Biochim.Biophys. Acta.; 1994; 1226:73-82.
- Bonilla E., Tanji K., Hirano M., Vu t.H., Dimauro s., schon E.A.; Mitochondrial involvement in Alzheimer’s disease. Biochim. Biophys. Acta; 1999; 1410:171-182.
- Boulet L., karpati G., shoubridge E.A.; Distribution an threshold expression of the tRNALys mutation in skeletal muscle of patients with myoclonic epilepsy and ragged- red fibers (MERRF). Am.j.Hum. Genet.; 1992; 51 :1187-1200.
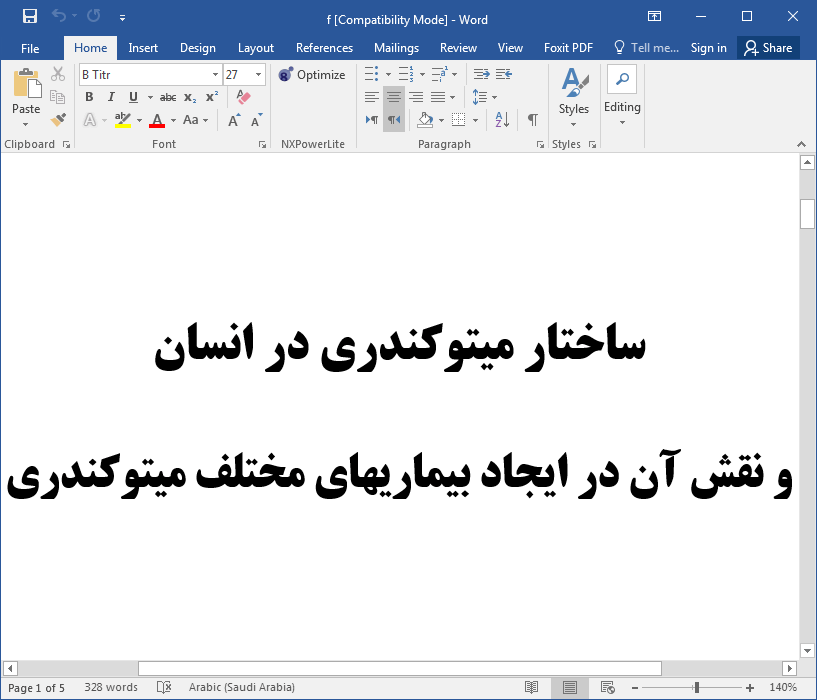
- Cann R.L., stoneking M., Wilson A.c.; Mitochondrial DNA and Human evolution. Nature; 1987;325:31-36.
- Case J.T.,Wallace D.C.; Maternal inheritance of mitochondrial DNA polymorphisms in cultured human fibroblasts. Somatic Cell Genetic; 1981;7:103-108.
- Carelli V.,Ghelli A.,RattaN.; Leber’s hereditary optic neuropathy: Biochemical effect of 11778/ND4 and 3460/ND1 mutations and correlation with the mitochondrial genotype. Neurology; 1997; 48:1623-1632.
- Chinnery P.F., Turnbull D.M.; Mitochondrial DNA disease. Lancet; 1999 ; 354: 17-21.
- David L.R imoin, Michael C.,Reed E.; EMERY and RIMOIN’S principles and practice of Medical Genetics. Fourth edition;2001.
- Dumoulin R.,Lopez R.F., Barker W.W.; A Novel gly 290 asp mitochondrial cytb mutation linked to a complex III deficiency in progressive exercise intolerance. Molecular and cellular probes; 1996 ; 10:389-391.
- Enriquez J.E., chomy A., and Attardi G.; MtDNA mutation in MERRF syndrome causes defective aminoacylation of tRNALys and premature translation termination. Nature. Genet.;1995; 10:
47-55. - Francesca O., Michele R., Anna M., Pietro T., Gabriella S.; Pathogenic role of mtDNA duplications in mitochondrial disease associated with mtDNA deletions. American Journal of Medical Genetics; 2003;118(3): 247-254.
- Houshmand M.;Mitochondrial DNA mutations, pathogenicity and inheritance. Iranian Journal of Biotechnology; 2003; vol.1; No.1.
- Herman A.C., Benttage and Giuseppe Attardi; Relationship of genotype to phenotype in fibroblast-derived transmitochondrial cell lines carrying the 3243 mutation associated with the MELAS encephalomyopathy: shift towards mutant genotype and role of mtDNA copy number. Human Molecular Genetics; 1996 ; 5(2):197-205.
- Josep L.; Pathology of the mitochondrion. Autonomous university of Barcelona, Barcelona, spain; 2003.
- Johns D.R.,Neufeld M.J., porrk R.D.; An ND mitochondrial DNA mutation associated with leber hereditary optic neuropathy. Biochem. Biophys. Res. Commun.; 1992a; 187:1551-1557.
- Kadowaki T., Sakura H., Otabe S.; Subtype of diabetes mellitus associated with a mutation in the mitochondrial gene. Muscle and Nerve; 1995;3:137-141.
- Kanamori A., Tanaka K., Umezawa S.; Insulin resistance in mitochondrial gene mutation. Diabetes care; 1994; 17: 778-779.
- King M.P., Koga Y.,Davidson M.,Schon E.A.; Defects in mitochondrial protein synthesis and respiratory chain activity segregate with the tRNALeu(uuR) mutation associated with mitochondrial myopathy, encephalopathy, Lactic acidosis, and stroke like episodes. Mol. Cell. Biol.; 1992; 12:480-490.
- Lu C.Y., Lee H.C.,Fahn H.J., Wei Y.H.; Oxidative damage elicited by imbalance of free radical scavenging. Enzymes is associated with large- scale mtDNA deletions in aging human skin- skin. Mutat,Res.; 1999; 25(2):11-21.
- Lenaz G.; Role of mitochondria in oxidative stress and aging Biochim. Biophys. Acta.; 1998; 1366: 53-67.
- Lee H.C., wei Y.H.; Mutation and oxidative damage of mtDNA and defective turnover of mitochondria in human aging. J.Formos. Med.Assoc.; 1997; 96:770-800.
- Lois A.,Tully C., Barbara C., Levin; Human mitochondrial Genetics. Biotechnology and Genetic Engineering Reviews; 2000; Vol.17.
- Lee H.C., Wei Y.H.; Mitochondrial alterations, cellular response to oxidative stress and defective degradation of proteins in aging. Biogerontology; 2001; 2(4): 231-240.
- Miyabayashi S.,Nakamura R., Tada K.; Defects of mitochondrial respiratory enzymes cloned cells from MELAS fibroblasts. J. Inher. Metab. Dis.; 1992;15:797-802.
- MtDNA in aging, cancer and mitochondrial disease. Innovita Research Foundation; 2003.
- MtDNA change during aging. Innovita ,Research Foundation; 2003.
- Rew D.A.; Mitochondrial DNA , human evolution and the cancer genotype. EJSO; 2001; 27:209-211.
- Rew D-A.; Tahe evolutionary biology of aging, death and human malignancy. Eur.J.Surg. Oncol.; 1998;24:568-72.
- Richardson A.; Increased oxidative damage is correlated to altered mitochondrial function in heterozygous manganese superoxide dismutase knockout mice. J.Biol. chem.; 1998; 237: 28510-28515.
- Schon E.A., Hirano M., Dimauro S.; Mitochondrial encephalomyopathies: clinical and Molecular analysis. J.Bioenerg. Biomem.; 1994; 26: 291-29.
- Sastre J., pallardo F.V., Vina J.; Mitochondrial oxidative stress plays a key role in aging and apoptosis. IUBMB Life; 2000; 49(5): 427-35.
- Sastre J.,Pallardo F.V., Garcia d.,Vina J.; Mitochondria, oxidative stress and aging. Free, Radic. Res; 2000; 32(3): 189-198.
- Wei Y.H., Lee H.C.; oxidative stress, mitochondrial DNA mutation and Impairment of antioxidant enzymes in aging. Exp. Biol. Med.; 2002; 227(9): 671-820.
- Wei Y.H., Ma Y.S., Lee H.C., Lu C.Y.; Mitochondrial theory of aging matures- roles of mtDNA mutation and oxidative stress in human aging. Zhonghua. Yi. Xue. Za. Zhi (taipei); 2001; 64(5):259-70.
- Wallac C.; Mitochondrial DNA mutations in diseases of energy metabolism. J.Bioenerg. Biomem; 1994;26 : 241-250.
- Wei Y.H.; oxidative stress and mitochondrial DNA mutations in human aging. Proc. Soc. Exp.Biol. Med.; 1998; 217(1):53-63.
- Wallace D.C.;Mitochondrial DNA sequence Variation in human evolution and disease. Proc. Natl. Acad. Sci. USA.; 1994;91:8739-8746.
- White F.A., Bunn C.L.; Segregation of mtDNA in Human somatic cell by hybrides. Mol. Gen. Genet.; 1984; 197; 453-46.
- Yoshino H.,Nakagawa Y., Kondo T.,mizuno Y.; Mitochondrial complex I and II activities of lymphocytes and platelets in parkinson’s disease. J. Neur., Trans. Parkinson’s. Dis. Dement. Sect.; 1992; 2: 27-34.
- Zeviani M., Muntoni F., Savarese N.;A MERRF MELAS overlap syndrome associated with a new point mutation in the mitochondrial DNA tRNA Lys Eur.J.Hum. Genet.; 1993; 1: 80-87.
- Zhang C., Baumer A., Maxwell R.J., Nagley p.; Multiple mtDNA deletions in an elderly human individual. FEBS; 1992:4-8.

- مقاله درمورد ساختار میتوکندری در انسان و نقش آن در ایجاد بیماری های مختلف میتوکندری
- پروژه دانشجویی ساختار میتوکندری در انسان و نقش آن در ایجاد بیماری های مختلف میتوکندری
- پایان نامه در مورد ساختار میتوکندری در انسان و نقش آن در ایجاد بیماری های مختلف میتوکندری
- تحقیق درباره ساختار میتوکندری در انسان و نقش آن در ایجاد بیماری های مختلف میتوکندری
- مقاله دانشجویی ساختار میتوکندری در انسان و نقش آن در ایجاد بیماری های مختلف میتوکندری
- ساختار میتوکندری در انسان و نقش آن در ایجاد بیماری های مختلف میتوکندری در قالب پاياننامه
- پروپوزال در مورد ساختار میتوکندری در انسان و نقش آن در ایجاد بیماری های مختلف میتوکندری
- گزارش سمینار در مورد ساختار میتوکندری در انسان و نقش آن در ایجاد بیماری های مختلف میتوکندری
- گزارش کارورزی درباره ساختار میتوکندری در انسان و نقش آن در ایجاد بیماری های مختلف میتوکندری